FMODB: The database of quantum mechanical data based on the FMO method
Last updated: 2025-09-03
All entries: 45600
Number of unique PDB entries: 8613
FMODB ID: M99KZ
Calculation Name: 6W63-AB-Docking457-pose4
Preferred Name:
Target Type:
Ligand Name: n-(4-tert-butylphenyl)-n-[(1r)-2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl]-1h-imidazole-4-carboxamide
Ligand 3-letter code: X77
Ligand of Interest (LOI): 1UN
PDB ID: 6W63
Chain ID: AB
UniProt ID: P0DTD1
Base Structure: Docking
Registration Date: 2024-01-29
Reference: Handa, Y.; Okuwaki, K.; Kawashima, Y.; Hatada, R.; Mochizuki, Y.; Komeiji, Y.; Tanaka, S.; Furuishi, T.; Yonemochi, E.; Honma, T.; Fukuzawa, K. Prediction of Binding Pose and Affinity of SARS-CoV-2 Main Protease and Repositioned Drugs by Combining Docking, Molecular Dynamics, and Fragment Molecular Orbital Calculations. J. Phys. Chem. B 2024, 128, 10, 2249-2265
Apendix: None
Modeling method
| Optimization | MOE:Amber10:EHT |
|---|---|
| Restraint | OptAll(Heavy atoms were optimized with constraints.) |
| Protonation | MOE:Protonate 3D |
| Complement | MOE:Structure Preparation |
| Water | No |
| Procedure | Manual calculation |
FMO calculation
| FMO method | FMO2-MP2/6-31G(d) |
|---|---|
| Fragmentation | Manual |
| Number of fragment | 309 |
| LigandResidueName | 1UN |
| LigandFragmentNumber | 306 |
| LigandCharge | |
| Software | ABINIT-MP - Open Ver. 1 Rev. 22 / 20200603(SMP) |
Total energy (hartree)
| FMO2-HF: Electronic energy | -4392590.00893 |
|---|---|
| FMO2-HF: Nuclear repulsion | 4268877.286569 |
| FMO2-HF: Total energy | -123712.722361 |
| FMO2-MP2: Total energy | -124060.845073 |
3D Structure
Ligand structure
1UN
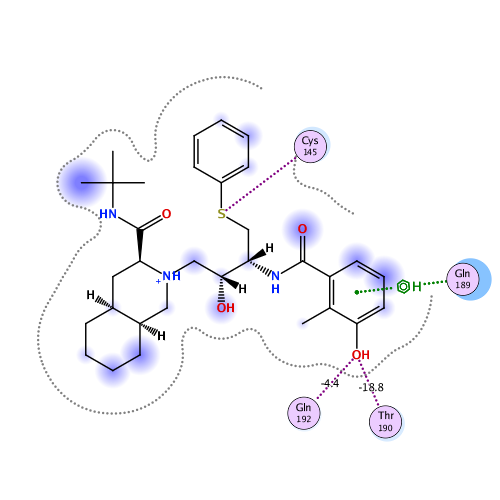
Ligand Interaction
Ligand binding energy (frag 306-309 : frag 1-305)
| IFIE [kcal/mol] | PIEDA [kcal/mol] | Charge transfer value [e] | |||
|---|---|---|---|---|---|
| IFIE SUMIFIE SUM at MP2 level. | ESElectro static interaction energy. | EXExchange-repulsion energy. | CT+mixCharge transfer and mixing terms energy. | DI(MP2)Dispersion energy. | q(I=>J)Charge transfer value from I to J fragments. |
| -174.3026 | -145.8603 | 89.8697 | -33.4269 | -84.8851 | 0.1365 |
Interactive mode: IFIE and PIEDA for fragment #306(B:201:1UN)
Summations of interaction energy for
fragment #306(B:201:1UN)
| IFIE [kcal/mol] | PIEDA [kcal/mol] | Charge transfer value [e] | |||
|---|---|---|---|---|---|
| IFIE SUMIFIE SUM at MP2 level. | ESElectro static interaction energy. | EXExchange-repulsion energy. | CT+mixCharge transfer and mixing terms energy. | DI(MP2)Dispersion energy. | q(I=>J)Charge transfer value from I to J fragments. |
Interaction energy analysis for fragmet #306(B:201:1UN)
| frag_NumFragment number. | ChainChain species. | Res #Residue number. | RES3-letter code of amino acid residue, ligand and solvent molecule. | FCHARGEFormal charge [e]. | q_MullikenFragment charge evaluated by Mulliken charge [e]. | q_NPAFragment charge evaluated by natural population analysis(NPA) charge [e]. | DISTDistance from Ligand [Å]. | TotalIFIE at MP2 level [kcal/mol]. | ESElectro static interaction energy by PIEDA [kcal/mol]. | EXExchange-repulsion energy by PIEDA [kcal/mol]. | CT+mixCharge transfer and mixing terms energy by PIEDA [kcal/mol]. | DI(MP2)Dispersion energy by PIEDA [kcal/mol]. | q(I=>J)Charge transfer value from I to J fragmens [e]. |
|---|